XAFSmass¶
- Release:
1.8.0

- Date:
Apr 17, 2026
- Authors:
Konstantin Klementiev (MAX IV Laboratory), Roman Chernikov (NSLS-II)
- License:
Open Source, MIT License
A program for calculating the mass of XAFS [X-ray Absorption Fine Structure] samples. The chemical formula parser understands parentheses and weight percentage, also in nested form. XAFSmassQt reports the quantity (weight, thickness or pressure) together with the expected height of the absorption edge.
Dependencies¶
numpy, matplotlib and pyparsing are required. Qt must be provided by either PyQt5, PySide2, PyQt6 or PySide6 by means of qtpy.
Get XAFSmass¶
XAFSmass is available as source distribution from PyPI or GitHub. The distribution archive also includes this documentation.
Running without installation¶
Unzip the .zip file from GitHub into a suitable directory and run
python XAFSmassQt.py. One advantage of no installation is a single location
of XAFSmass served by any Python installation.
Running with installation¶
From the unzipped directory that has pyproject.toml run
python -m pip install . or run pip install xafsmass to get it directly
from PyPI. After installation, XAFSmass can be started by xafsmass command.
Citing XAFSmass¶
Please cite XAFSmass as: K. Klementiev and R. Chernikov, “XAFSmass: a program for calculating the optimal mass of XAFS samples”, J. Phys.: Conf. Ser. 712 (2016) 012008, doi:10.1088/1742-6596/712/1/012008.
Theoretical references used¶
The tabulated scattering factors are taken from Henke et al. (10 eV < E < 30 keV) [Henke], Brennan & Cowan (30 eV < E < 509 keV) [BrCo] and Chantler (11 eV < E < 405 keV) [Chantler].
Note
The tables of f’’ factors consider only photoelectric cross-sections. The tabulation by Chantler can optionally have total absorption cross-sections. This option is enabled by selecting the data table ‘Chantler total (NIST)’.
http://henke.lbl.gov/optical_constants/asf.html B.L. Henke, E.M. Gullikson, and J.C. Davis, X-ray interactions: photoabsorption, scattering, transmission, and reflection at E=50-30000 eV, Z=1-92, Atomic Data and Nuclear Data Tables 54 (no.2) (1993) 181-342.
http://www.bmsc.washington.edu/scatter/periodic-table.html ftp://ftpa.aps.anl.gov/pub/cross-section_codes/ S. Brennan and P.L. Cowan, A suite of programs for calculating x-ray absorption, reflection and diffraction performance for a variety of materials at arbitrary wavelengths, Rev. Sci. Instrum. 63 (1992) 850-853.
http://physics.nist.gov/PhysRefData/FFast/Text/cover.html http://physics.nist.gov/PhysRefData/FFast/html/form.html C. T. Chantler, Theoretical Form Factor, Attenuation, and Scattering Tabulation for Z = 1 - 92 from E = 1 - 10 eV to E = 0.4 - 1.0 MeV, J. Phys. Chem. Ref. Data 24 (1995) 71-643.
Usage¶
Chemical formula parser¶
The parser understands chemical elements, optionally followed by atomic quantities or weight percentages. A group of atoms can be enclosed in parentheses and assigned a common quantity or wt%. Some examples are given above the edit line. For example, Cu%1Zn%1((Al2O3)%10SiO2) means 1 wt% of Cu and 1 wt% of Zn in an aluminosilicate matrix composed of 10 wt% of alumina in silica.
For the search of an unknown elemental concentration, give x to the element of interest.
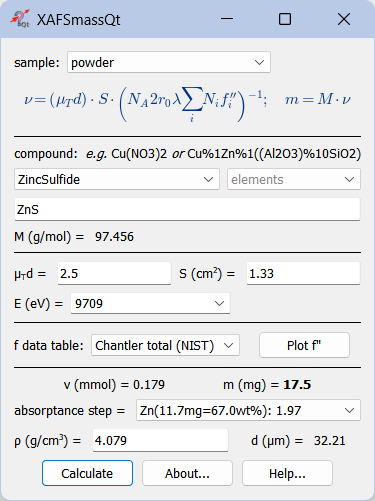
Calculation of mass and absorption step for powder samples¶
Note
You typically do not need the calculated values at exactly the edge position but rather at an energy somewhere above it. The list of edges offers the edge positions plus 50 eV. You are free to specify any energy within the range of the selected tabulation.
The most common use is determining the mass of a powder sample. The optimal optical thickness μd depends on the absorption levels chosen for the ionization chambers (see below). In practice, μd typically lies between 2 and 3. For example, with a 17.4% absorption in the first chamber and 50% in the second, the optimal thickness is 2.42. If the absorption step exceeds 1.5 (as reported by the drop‑down menu “absorptance step =”), the sample mass should be reduced to avoid the potential thickness effect arising from possible inhomogeneities in the wafer. If the sample is diluted and the absorption step is very low, increasing the wafer thickness will not improve the spectra. The optimal thickness already provides the best signal‑to‑noise ratio. In such cases, improved results can only be obtained by using an alternative detection mode, such as fluorescence or electron yield measurements.
|
|
Calculation of thickness and absorption step for samples with known density¶
Here you can calculate the thickness of a sample with known density, typically a foil. Commercial foils are generally highly uniform in thickness, so large absorption steps and the potential thickness effect can be neglected.
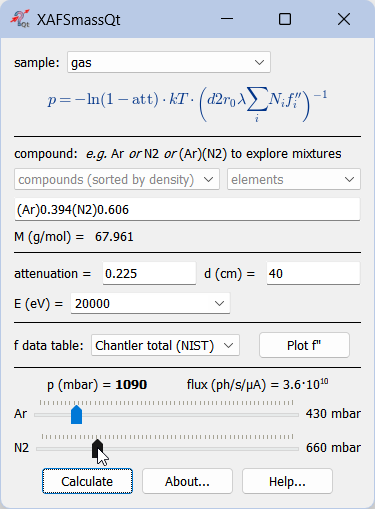
Calculation of gas pressure for ionization chambers¶
Caution
For nitrogen, do not forget the 2: N2, not just N!
Start with the 2nd ionization chamber (IC). If a reference foil is placed between the 2nd and the 3rd IC, the fraction of x-rays absorbed by the 2nd IC is usually set to 50%. If the reference foil is not needed, one can select the total absorption (close to 100%). For these two cases the optimal absorption of the 1st IC at a certain μd is found from the figures above showing the levels of signal-to-noise ratio.
For exploring mixtures of several gases, give the gases in parentheses, e.g. as (Ar)(N2). Each gas will get a slider defining its partial pressure. The program will calculate the molar weight of each gas and update the chemical formula and the total attenuation.
Absolute flux is reported per current unit of the ionization chamber. This calculation needs electron-ion pair energy that is taken from xdb.lbl.gov/Section4 where it is given for a few common gases.
Calculation of unknown elemental concentration¶
Case 1: You know the composition of the matrix¶
You need an absorption spectrum taken without the sample (an empty spectrum) obtained with the same state of the ionization chambers. Subtract this spectrum from the sample spectrum to obtain the true absorption coefficient (without any vertical offset).Then determine the value of μd above the edge (μTd), the edge jump (Δμd) and its uncertainty (δμd). Specify the chemical formula with x.
Case 2: You know the sample mass and area¶
Determine the edge jump (Δμd). For the pure element, adjust μTd until the absorption step shown in the pull‑down list matches the experimentally measured Δμd. This yields the mass of the element of interest. Dividing this value by the total sample mass gives the corresponding weight percentage.
Finding scattering factors f’’¶
If you need to know the scattering factor f’’ at different energies and/or its jump at an edge (Δf’’), XAFSmass provides a graphical tool for this.
For example, you may need these values to determine the composition of a binary compound if you have the experimental edge heights at two edges. The absorption step Δμd at an absorption edge of energy E is proportional to Δf’’ν/E, where ν is the amount of (resonantly) absorbing atoms in mole. Hence, the atomic ratio of two elements in the same sample is \(\nu_A/\nu_B = (\Delta\mu d)_A/(\Delta\mu d)_B\cdot[\Delta f_B'' /\Delta f_A'' \cdot E_A/E_B]\). For binary compounds \({\rm A}_x{\rm B}_{1-x}\) the concentration \(x\) is calculated as \(x = (\nu_A/\nu_B)/[1+(\nu_A/\nu_B)]\).